Simulazione di Dinamica Molecolare al Supercalcolatore ENEA-CRESCO per identificare le interazioni tra PM2,5 e il virus SARS-CoV-2
Il fortissimo eccesso di mortalità registrato al Nord Italia, nelle regioni della Pianura Padana durante i mesi invernali della “prima ondata” e delle ondate successive, mantiene ancora acceso a tutt’oggi il dibattito sulla possibile azione del particolato atmosferico quale carrier/vettore nella trasmissione aerodispersa del virus SARS-CoV-2.
Tra i diversi aspetti affrontati dal progetto PULVIRUS c’è anche quello di delucidare la possibile associazione tra il particolato atmosferico e il bioaerosol attraverso il quale si trasferisce il virus SARS-CoV-2. Per rispondere a questa domanda, in Pulvirus si è adottato anche l’approccio in silico, qui descritto con i primi modelli strutturali ottenuti mediante l’uso della modellistica molecolare e simulazioni di dinamica molecolare con calcolo ad alte prestazioni (HPC).
Il vantaggio degli esperimenti in silico risiede nella loro capacità di fornire una risoluzione spazio-temporale della biochimica delle interazioni difficilmente accessibile con l’uso di tecniche sperimentali [1,2]. Se queste tecniche hanno trovato, in seguito allo scoppio della pandemia da SARS-CoV-2, un largo impiego nella progettazione di farmaci terapeutici e vaccini [3,4] e se la quasi totalità degli studi di modellistica molecolare per SARS-CoV-2 è orientata alla verifica dell’interazione del virus con le strutture della cellula ospite [5], nessuna informazione riguardo una possibile interazione con le componenti dell’aerosol è riportata ad oggi in letteratura.
In questa newsletter presentiamo la strategia in silico adottata in Pulvirus per calcolare la forza delle potenziali interazioni inter-molecolari tra le componenti dell’aerosol atmosferico e le proteine strutturali di superficie di SARS-CoV-2, con lo scopo ultimo di attestare l’eventuale capacità da parte del particolato atmosferico (PM) di riconoscere e trasportare in maniera stabile il virus aerodisperso. Nonostante le attuali tecniche computazionali consentano, in principio, di modellare e simulare l’intero processo di riconoscimento tra frammenti anche molto grandi di PM e di virus, questo processo rimane ancora un lavoro estremamente complesso che richiede tempi e costi computazionali elevati. Per ridurre il costo computazionale ed accorciare i tempi di calcolo sono stati realizzati modelli semplificati del PM2.5 e della componente biologica di una possibile interfaccia PM-virus che sarà oggetto di analisi di dinamica molecolare.
Per la componente biologica dell’interfaccia, è stato realizzato un frammento della membrana lipidica dell’involucro virale nella quale sono stati inseriti i modelli strutturali delle proteine di superficie: la glicoproteina spike (S), la proteina di membrana (M) e la proteina di rivestimento (E) (Figura 1). La costruzione del modello preliminare strutturale è stata realizzata tenendo conto della topologia assunta dalle diverse proteine nella membrana virale, dalla loro capacità di formare oligomeri e sfruttando approcci che si basano sulla omologia di sequenza e metodologie template-free o ab-initio [6-8].
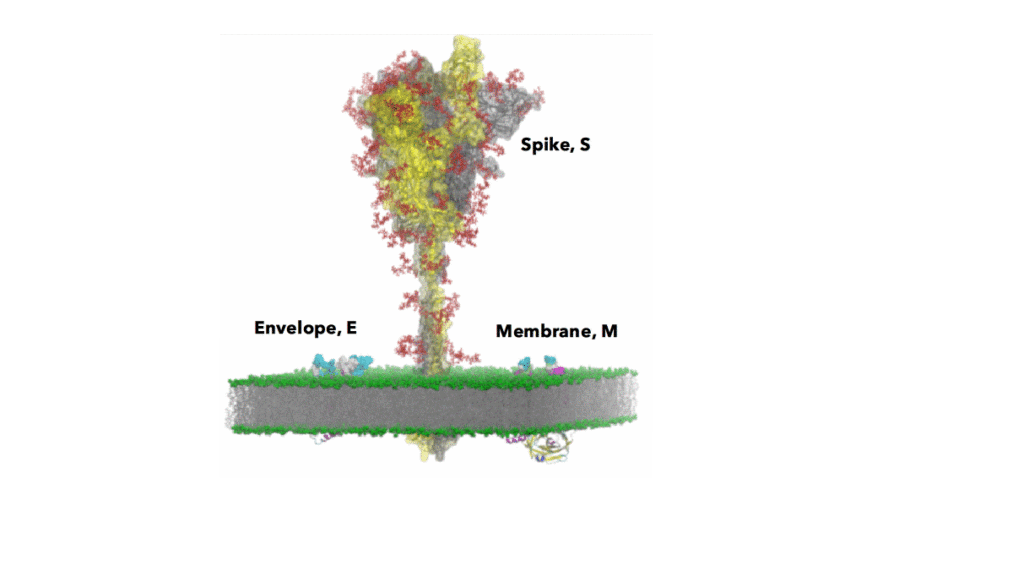
Figura 1
Modello strutturale di un frammento del virus SARS-CoV-2. Da sinistra la proteina E nella forma pentamerica, al centro l’ intera proteina S glicosilata con il dominio RBD nella conformazione aperta e a destra la proteina M nella forma omodimerica. Le tre catene individuali della proteina S sono colorate in giallo, grigio e bianco rispettivamente, mentre i glicani sono colorati in rosso. Le tre proteine strutturali di SARS-CoV-2 sono inserite nella membrana virale. I gruppi polari e le catene idrofobiche della membrana virale sono colorati in verde e grigio rispettivamente.
La costruzione del modello strutturale del PM2.5 ha richiesto uno sforzo di modellazione maggiore. Infatti sebbene in letteratura siano riportati alcuni modelli e simulazioni di dinamica molecolare di aerosol primario, quasi tutti focalizzati sul processo di nucleazione [9-15], solo scarsissime informazioni sono disponibili riguardo a modelli strutturali di aerosol secondario [16-17]. Un’ulteriore complicazione risiede nel fatto che i pochi modelli realizzati in questi lavori e i relativi parametri da inserire nei force field dei codici di calcolo non sono trasferibili, a meno di consistenti modifiche, nei codici utilizzati per simulare anche la componente biologica dell’interfaccia. Il modello strutturale preliminare di un frammento di PM2.5, qui riportato, è stato ottenuto mediante simulazioni di dinamica molecolare e utilizzando le risorse computazionali della infrastruttura ENEA – CRESCO/ENEAGRID High Performance Computing Infrastructure [16]. In breve, a partire dalle varie componenti organiche ed inorganiche che compongono un tipico aerosol secondario delle zone maggiormente inquinate del Nord Italia, è stato realizzato un sistema in cui le molecole organiche ed inorganiche sono state disposte in maniera casuale in prossimità di un frammento di origine carboniosa e ad una tipica concentrazione registrata durante un picco di inquinamento [18]. Il sistema di coordinate atomiche ottenuto è stato sottoposto a simulazioni di dinamica molecolare (MD). Durante 200 ns di simulazione (288 core, circa 5 mesi di tempo di calcolo) è possibile osservare fenomeni di aggregazione (clusters) delle molecole organiche del PM e della loro migrazione verso la componente carboniosa con la quale interagiscono stabilmente; la componente inorganica del PM, come atteso, rimane in forma solubile (Figura 2, ).
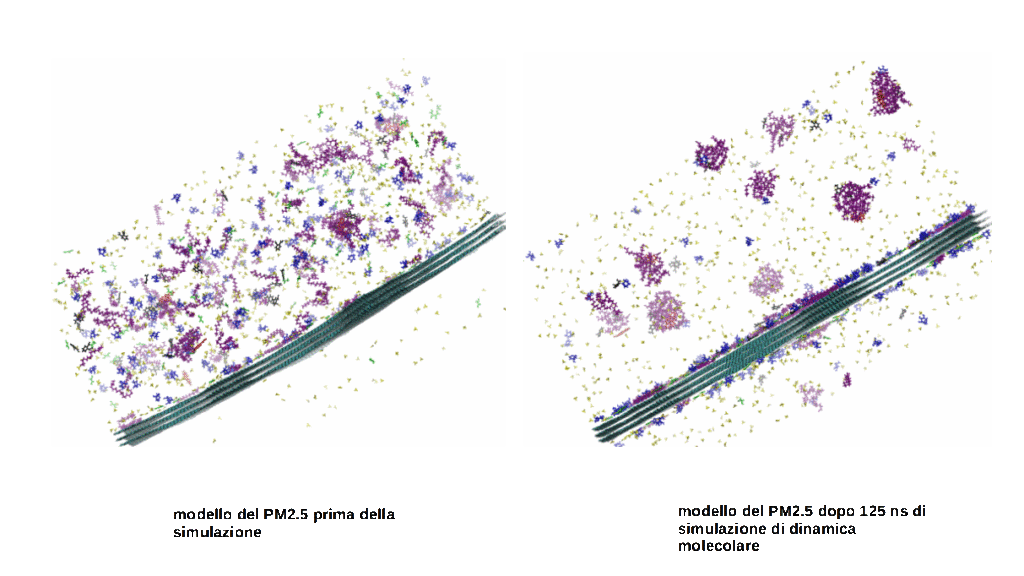
Figura 2
Modello strutturale di un frammento di aerosol secondario all’inizio (a sinistra) e dopo 125 ns di simulazione di dinamica molecolare (a destra). La componente carboniosa è rappresentata da un foglietto di grafene (celeste). Durante la simulazione, gli idrocarburi aromatici policiclici (rosso), gli zuccheri anidri (porpora), gli acidi n-alcaloidi (blu), gli acidi policarbossilici (nero) aromatici, gli acidi dicarbossilici aromatici (verde), che nell’insieme rappresentano la componente organica secondaria del PM, tendono ad aggregarsi tra di loro in virtù delle loro proprietà chimico fisiche e ad avvicinarsi e interagire con la componente carboniosa del PM. La componente inorganica secondaria del PM (giallo) rimane insolubile.
I modelli semplificati del PM e della componente biologica saranno utilizzati per simulare le interazioni all’interfaccia PM-virus. I risultati di questi studi, attesi nei prossimi mesi, consentiranno di comprendere non solo i meccanismi di riconoscimento, se presenti, tra le proteine di superficie del virus e le componenti organiche ed inorganiche del aerosol ma anche i possibili effetti del PM sulla integrità del virus e in particolare delle glicoproteine di membrana che hanno un ruolo fondamentale nel riconoscimento della cellula ospite.
Per maggiori informazioni, si rimanda ai report dell’attività 5.1 dell’obiettivo 5 del Progetto Pulvirus https://www.pulvirus.it/index.php/documentazione-obiettivo-5/
Alla stesura della newsletter hanno contribuito tutti i ricercatori ENEA che hanno partecipato all’attività 5.1 dell’Obiettivo 5.
Per maggiori informazioni contattare
Caterina Arcangeli caterina.arcangeli@enea.it
Bibliografia:
- Ozboyaci, M., Kokh, D.B., Corni, S., Wade, R.C., 2016. Modeling and simulation of protein–surface interactions: achievements and challenges. Quarterly Reviews of Biophysics 49. https://doi.org/10.1017/S0033583515000256
- Hollingsworth, S.A., Dror, R.O., 2018. Molecular Dynamics Simulation for All. Neuron 99, 1129–1143. https://doi.org/10.1016/j.neuron.2018.08.011
- European Commission – Exscalate4Cov project : supercomputing to identify new therapies for COVID-19 https://digital-strategy.ec.europa.eu/en/policies/high-performance-computing
- https://www.dompe.com/en/media/press-releases/leonardo-e-domp%C3%A9-nasce-la-prima-infrastruttura-nazionale-di-sicurezza-sanitaria-digitale-con-architettura-cloud
- Amaro, R.E., Mulholland, A.J., 2020. Biomolecular Simulations in the Time of COVID-19, and After. Computing in Science & Engineering 22, 30–36. https://doi.org/10.1109/MCSE.2020.3024155
- Mahtarin, R., Islam, S., Islam, M.J., Ullah, M.O., Ali, M.A., Halim, M.A., 2020. Structure and dynamics of membrane protein in SARS-CoV-2. Journal of Biomolecular Structure and Dynamics 0, 1–14. https://doi.org/10.1080/07391102.2020.1861983
- Mandala, V.S., McKay, M.J., Shcherbakov, A.A., Dregni, A.J., Kolocouris, A., Hong, M., 2020. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nature Structural & Molecular Biology 27, 1202–1208. https://doi.org/10.1038/s41594-020-00536-8
- Woo, H., Park, S.-J., Choi, Y.K., Park, T., Tanveer, M., Cao, Y., Kern, N.R., Lee, J., Yeom, M.S., Croll, T.I., Seok, C., Im, W., 2020. Developing a Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein Model in a Viral Membrane. J. Phys. Chem. B 124, 7128–7137. https://doi.org/10.1021/acs.jpcb.0c04553
- Hede, T., Li, X., Leck, C., Tu, Y., Ågren, H., 2011. Model HULIS compounds in nanoaerosol clusters – investigations of surface tension and aggregate formation using molecular dynamics simulations. Atmospheric Chemistry and Physics 11, 6549–6557. https://doi.org/10.5194/acp-11-6549-2011
- Mao, Q., van Duin, A.C.T., Luo, K.H., 2017. Formation of incipient soot particles from polycyclic aromatic hydrocarbons: A ReaxFF molecular dynamics study. Carbon 121, 380–388. https://doi.org/10.1016/j.carbon.2017.06.009
- Mansurov, U., Shah, D., Bazybek, N., Amouei Torkmahalleh, M., 2018. Particulate Matter Formation in Post-combustion CO2 Capture Columns: Insights from Molecular Dynamics Simulations. Energy Fuels 32, 12679–12688. https://doi.org/10.1021/acs.energyfuels.8b02647
- Karadima, K.S., Mavrantzas, V.G., Pandis, S.N., 2019. Insights into the morphology of multicomponent organic and inorganic aerosols from molecular dynamics simulations. Atmospheric Chemistry and Physics 19, 5571–5587. https://doi.org/10.5194/acp-19-5571-2019
- Roose, A., Toubin, C., Dusanter, S., Riffault, V., Duflot, D., 2019. Classical Molecular Dynamics Study of Small-Chain Carboxylic Acid Aerosol Particles. ACS Earth Space Chem. 3, 380–389. https://doi.org/10.1021/acsearthspacechem.8b00172
- Violi, A., Venkatnathan, A., 2006. Combustion-generated nanoparticles produced in a benzene flame: A multiscale approach. J. Chem. Phys. 125, 054302. https://doi.org/10.1063/1.2234481
- Zhou, X., Zhou, Y., Picaud, S., Devel, M., Carrete, J., Madsen, G.K.H., Wang, Z., 2020. Role of black carbon in the formation of primary organic aerosols: Insights from molecular dynamics simulations. Atmospheric Chemistry and Physics Discussions 1–17. https://doi.org/10.5194/acp-2020-81
- Roston, D., 2021. Molecular dynamics simulations demonstrate that non-ideal mixing dominates subsaturation organic aerosol hygroscopicity. Phys. Chem. Chem. Phys. 23, 9218–9227. https://doi.org/10.1039/D1CP00245G
- Darvas, M., Picaud, S., Jedlovszky, P., 2010. Molecular Dynamics Simulation of the Adsorption of Oxalic Acid on an Ice Surface. ChemPhysChem 11, 3971–3979. https://doi.org/10.1002/cphc.201000513
- Iannone, F., Ambrosino, F., Bracco, G., De Rosa, M., Funel, A., Guarnieri, G., Migliori, S., et al. 2019. “CRESCO ENEA HPC Clusters: A Working Example of a Multifabric GPFS Spectrum Scale Layout.” In 2019 International Conference on High Performance Computing and Simulation, HPCS 2019, 1051–52. Institute of Electrical and Electronics Engineers Inc. https://doi.org/10.1109/HPCS48598.2019.9188135.
- Comunicazione interna: Gualtieri, M., Straquadanio, M., Malaguti, A. 2021